Las enfermedades venéreas constituyen una de las principales causas infecciosas de pérdidas reproductivas en bovinos, particularmente en sistemas de cría donde el servicio natural continúa siendo la modalidad predominante. A diferencia de otras patologías reproductivas, estas infecciones suelen cursar de forma subclínica en los toros, que actúan como portadores asintomáticos y reservorios del agente, favoreciendo su diseminación dentro del rodeo.
La identificación precisa de los agentes etiológicos involucrados en abortos resulta fundamental para la implementación de medidas sanitarias adecuadas. En este contexto, Campylobacter fetus representa un desafío diagnóstico, ya que sus dos principales subespecies: C. fetus subsp. venerealis (CFV) y C. fetus subsp. fetus (CFF), presentan alta similitud genética, pero difieren en su epidemiología y en su impacto sanitario.
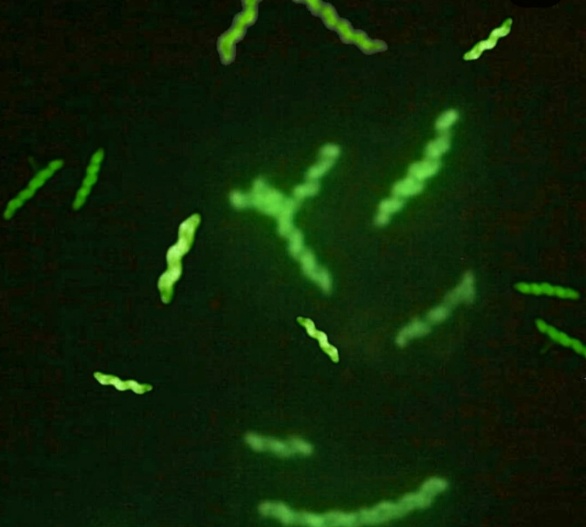
En Laboratorio 9 de Julio se recibieron dos fetos abortados: uno bovino, proveniente de Los Toldos en el mes de abril y otro ovino, provenientes de 25 de Mayo en Julio. A partir de estas muestras se logró el aislamiento de Campylobacter fetus mediante cultivo bacteriológico. La identificación se confirmó mediante inmunofluorescencia directa (Imagen 1) y técnicas moleculares (qPCR).
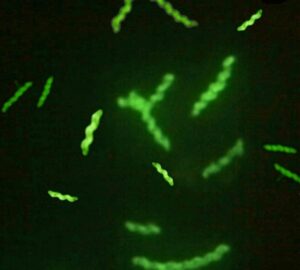 (Imagen 1. Inmunofluorescencia directa para Campylobacter fetus.)
(Imagen 1. Inmunofluorescencia directa para Campylobacter fetus.)
Mediante qPCR específica, se determinó que el aislamiento proveniente del feto bovino correspondía a C. fetus subsp. Venerealis (Imagen 2 y 3), mientras que el aislamiento ovino fue identificado como C. fetus subsp. fetus.
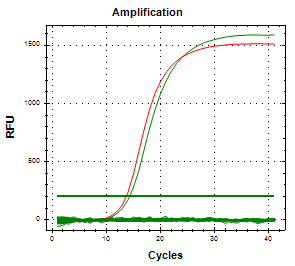 (Imagen 2. Curva de amplificación de CFV en feto bovino. Control positivo en rojo; muestra en verde.)
(Imagen 2. Curva de amplificación de CFV en feto bovino. Control positivo en rojo; muestra en verde.)
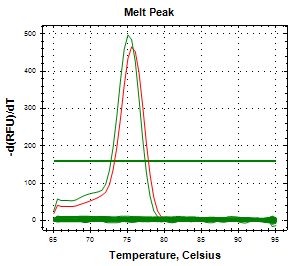 (Imagen 3. Curva de melting de CFV en feto bovino. Control positivo en rojo; muestra en verde.)
(Imagen 3. Curva de melting de CFV en feto bovino. Control positivo en rojo; muestra en verde.)
Como parte de la investigación epidemiológica posterior al diagnóstico del aborto bovino, se analizaron 12 toros del rodeo reproductivo, detectándose dos animales positivos a C. fetus subsp. venerealis, lo que confirma la circulación del agente en el establecimiento y su probable rol en las fallas reproductivas observadas.
Dado que la diferenciación entre subespecies puede resultar compleja mediante metodologías convencionales, se procedió a la secuenciación del genoma completo de los aislamientos. El ADN bacteriano fue enviado a la empresa Macrogen (Corea del Sur), donde se realizó secuenciación de alta calidad.
El análisis bioinformático de los datos permitió confirmar los resultados diagnósticos obtenidos. Los genomas fueron comparados con secuencias de referencia internacionales y se construyó un árbol filogenético (Imagen 4), en el cual ambos aislamientos se agruparon de manera consistente con sus respectivas subespecies.
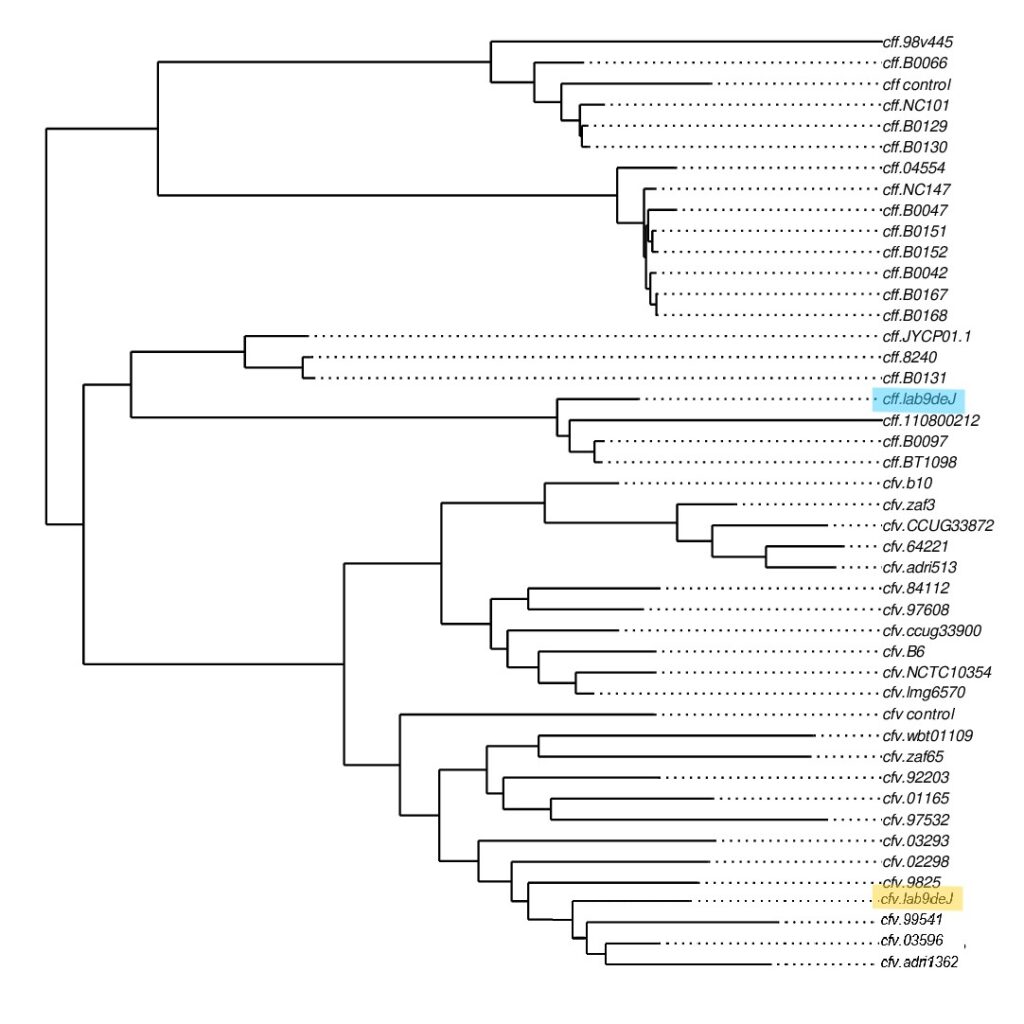 (Imagen 4. Árbol filogenético. En azul, aislamiento identificado como CFF; en amarillo, aislamiento identificado como CFV.)
(Imagen 4. Árbol filogenético. En azul, aislamiento identificado como CFF; en amarillo, aislamiento identificado como CFV.)
Este estudio pone de manifiesto la confiabilidad de las técnicas moleculares utilizadas en el diagnóstico de rutina y destaca la importancia de integrar los resultados de laboratorio con la información epidemiológica del establecimiento.
Si bien la secuenciación de genoma completo no constituye una herramienta de uso rutinario, su aplicación resulta de gran valor en la validación de diagnósticos complejos y en la caracterización de agentes involucrados en pérdidas reproductivas.
La incorporación de estas herramientas fortalece la capacidad diagnóstica y contribuye a la toma de decisiones sanitarias basadas en evidencia, orientadas a mejorar la eficiencia reproductiva de los sistemas productivos.
Lic. Pablo Addamo
paddamo@lab9dejulio.com.ar
M.V Clara Giovanini
clgiovanini@lab9dejulio.com.ar